Neste mês de fevereiro esta iniciando uma Pesquisa Clinica inédita no Brasil, o Registro Nacional de Acromegalia coordenado pela Sociedade Brasileira de Endocrinologia e Metabologia. Em Santa Catarina o Centro de Endocrinologia e Diabetes de Joinville – Endoville foi o primeiro centro aprovado em Santa Catarina para participar da Pesquisa, apoiado pelo Clube da Hipófise de Joinville.
Acromegalia é uma doença causada pelo excesso de produção do hormônio de crescimento (GH). O nome acromegalia é reservado à produção excessiva de GH na vida adulta, quando as cartilagens de crescimento já se encontram fechadas, inativas, causando aumento das mãos e dos pés. Quando o excesso de GH ocorre na infância ou na puberdade, antes do fechamento dessas cartilagens, a ação do GH provoca crescimento excessivo na estatura, e o quadro recebe o nome de Gigantismo.
O Registro Nacional de Acromegalia é um estudo aberto a todos os pacientes portadores da doença, com o objetivo de alertar e orientar sobre os sintomas e os tratamentos da doença, e estabelecer diretrizes de Saúde Pública para o tratamento da doença e de suas importantes comorbidades. As consultas podem ser agendadas gratuitamente pelo fone (47) 3029 5258.
Os principais sintomas são dores de cabeça, dores articulares, formigamentos, espaçamento entre os dentes, lábios grossos e alargamento do nariz. E as conseqüências mais comuns são diabetes, hipertensão, insuficiência cardíaca, aumento da tireóide e próstata, entre outras complicações, além da redução em média de 10 anos de vida.
INFORMAÇÕES ADICIONAIS SOBRE A DOENÇA
Funções do hormônio do crescimento (GH)
Produzido pela hipófise, o GH promove o crescimento de quase todas as células e tecidos do corpo humano.
Causas
Acromegalia e gigantismo são causados pela produção exagerada de GH. Em 98% dos casos, a produção excessiva está associada à presença de tumores benignos da hipófise: os adenomas. Os 2% restantes compreendem casos raríssimos de tumores malignos, ou ainda de tumores produtores de GH localizados no pâncreas, pulmões ou em outros tecidos. Existem, ainda, alguns casos raros de formas familiares da doença.
Prevalência
A doença é rara. Calcula-se que ocorram 50 a 70 casos por milhão de pessoas. Mulheres e homens são acometidos na mesma proporção.
Sintomas
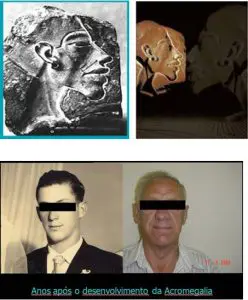
A acromegalia pode causar mudanças na aparência física, mas como a doença costuma evoluir no decorrer de vários anos, seus sinais podem ser confundidos com os do envelhecimento. Muitas vezes, o diagnóstico é feito casualmente por um médico que reconhece o aspecto físico do portador da doença num ambiente público ou quando se compara a fotografia atual do paciente com uma antiga.
As características mais importantes da aparência física são: 1) mãos e pés crescem, os sapatos não servem mais, os anéis não entram nos dedos; 2) alargamento da região frontal e da testa; 3) o queixo fica proeminente, dando ao rosto um aspecto característico; 4) espaçamento entre os dentes e perda dentária; 5) aumento do volume do tórax, do nariz e dos genitais; 6) os lábios engrossam.
Como o GH age em todos os tecidos do corpo, a produção excessiva afeta múltiplas funções orgânicas, provocando as seguintes alterações:
- A pele se torna espessa, oleosa, propensa à acne, deformada pelo aparecimento de pregas . Os pacientes apresentam suor excessivo;
- Alterações respiratórias: obstrução de vias aéreas, aumento das dimensões da língua, dificuldade de respiração durante o sono e espessamento das cordas vocais que tornam a voz mais grave;
- Alterações cardiovasculares: aumento do volume do coração, hipertensão arterial, insuficiência cardíaca, arritmias, fadiga;
- Alterações gastrointestinais: aumento de volume do fígado, pâncreas e outros órgãos. Pode haver formação de pólipos intestinais;
- Alterações metabólicas/endócrinas: diabetes, intolerância a carboidratos, resistência à insulina, aumento dos níveis de colesterol e dos triglicérides, nódulos na tireóide, diminuição do apetite sexual, saída de leite pelos mamilos da mulher e alterações menstruais;
- Alterações músculo-esqueléticas: dores articulares, artrite, formigamentos e alterações de sensibilidade da pele, síndrome do túnel do carpo, osteopenia (diminuição da massa óssea) e osteoporose.
- Alterações neurológicas: dor de cabeça persistente;
- Alterações oftálmicas: distúrbios visuais e redução do campo visual.
Diagnóstico
Quando a aparência física e o quadro clínico sugerem acromegalia, o diagnóstico pode ser confirmado por dois exames de sangue: as dosagens de GH e de IGF-1(fator de crescimento 1). Um único exame alterado de GH não permite fechar o diagnóstico, porque a hipófise libera esse hormônio de forma irregular, no decorrer do dia. Para obter dados mais confiáveis, os médicos fazem um tipo de exame de sangue no qual o GH é dosado depois da ingestão de carboidrato (curva glicêmica). Os níveis de IGF-1 são bem mais constantes no decorrer do dia. Valores elevados quase sempre estão associados à acromegalia.
Depois que o diagnóstico foi estabelecido, uma série de exames de imagem (tomografia computadorizada, ressonância nuclear magnética, etc.) é realizada para detectar a presença de tumores na hipófise ou em localizações mais raras.
Tratamento
Os objetivos do tratamento são:
Reduzir as concentrações de GH e de IGF-1 para os níveis normais;
Aliviar a pressão que tumores situados na hipófise possam exercem sobre o nervo óptico e áreas cerebrais vizinhas;
Preservar as funções da hipófise;
Reverter ou melhorar os sinais e os sintomas da acromegalia.
As opções de tratamento incluem:
Cirurgia
A cirurgia tem índices de cura de 80% a 90% nos casos em que os adenomas (tumores benignos) medem menos de 1 cm. Quando ultrapassam essa dimensão, os índices caem para menos de 50%.
Quando a cirurgia é realizada com sucesso, a aparência facial e o inchaço dos tecidos começam a melhorar em poucos dias. Mesmo assim, os níveis hormonais podem não retornar ao normal e exigir tratamento complementar com medicamentos.
A principal complicação cirúrgica é a lesão do tecido hipofisário normal, que pode exigir reposição hormonal para o resto da vida.
Radioterapia
É consenso entre os especialistas que a radioterapia NÃO deve ser usada como primeira opção de tratamento, exceto quando os tumores não podem ser removidos cirurgicamente, quando a doença persiste depois da operação, quando o tratamento com medicamentos falha ou quando os pacientes recusam outras opções.
Depois do tratamento radioterápico, a função hipofisária costuma declinar gradativamente. Depois de 10 anos, 70% dos casos necessitam de reposição hormonal prolongada.
Medicamentos
Três tipos de medicamentos são empregados no tratamento da acromegalia:
1 – cabergolina / bromocriptina: têm eficácia em apenas 10% dos casos. São usados preferencialmente quando os níveis de prolactina estão aumentados;
2 – octreotida / lanreotida: constituem o tratamento clínico de primeira linha, uma vez que 96% dos tumores que secretam GH apresentam em suas células receptores um fator de crescimento celular conhecido como somatostatina. Como as drogas deste grupo ligam-se a esses receptores, são dotadas da propriedade de regular a produção de GH e o crescimento celular.
Nos últimos 10 anos, a aplicação de octreotida por via intramuscular tornou-se a forma de tratamento clínico da acromegalia mais prescrita e mais estudada.
A administração de octreotida além de reduzir os níveis de GH e de IGF-1, diminui os índices de mortalidade, normaliza os níveis hormonais em 50% a 60% dos casos, diminui a freqüência dos batimentos cardíacos, melhora a função dos ventrículos, a resistência à atividade física e faz regredirem os sinais e sintomas da acromegalia;
3 – pegvisomant: são medicamentos úteis quando existir resistência aos anteriores. Embora normalizem os níveis de IGF-1 em cerca de 95% dos casos, seu efeito sobre o crescimento dos tumores hipofisários é questionável.
Considerações finais
Pacientes portadores de acromegalia que evoluem sem tratamento apresentam mortalidade mais elevada. Quanto mais altos os níveis de GH e de IGF-1, maior a probabilidade de surgirem complicações e mais altas as taxas de mortalidade.
O tempo de duração dos sintomas antes do diagnóstico, a duração da doença, a presença de diabetes, de doença cardiovascular e de hipertensão são fatores associados à mortalidade.
Por essa razão, é muito importante conhecer as características da doença para identificá-la o mais cedo possível e iniciar o tratamento antes que surjam complicações irreversíveis.
Mais informações:
47 – 3029 5258
Coordenação:
Dr. Luiz Antônio de Araújo – Investigador Principal.
Diretor do Departamento de Neuroendocrinologia da Sociedade Brasileira de Endocrinologia e Metabologia – SBEM; Diretor Técnico da Endoville; Presidente do Clube da Hipófise de Joinville.
Dra. Julia Appel – Sub Investigador
Corpo Clínico da Endoville; Diretora do Clube da Hipófise de Joinville.